流行病学
流行病学
流行病学:青年人中的成年发病型糖尿病(maturity-onset diabetesof the young,MODY)是一组具有高度遗传及临床表型异质性的单基因疾病,其特征为发病年龄小于25岁,有3代以上
糖尿病家族遗传史,符合常染色体显性遗传规律,以胰岛素分泌不足为原发病理基础,属于胰岛β细胞功能遗传性缺陷的特殊类型。
青年人中的成年发病型糖尿病(maturity-onset diabetesof the young,MODY)是一组具有高度遗传及临床表型异质性的单基因疾病,其特征为发病年龄小于25岁,有3代以上
糖尿病家族遗传史,符合常染色体显性遗传规律,以胰岛素分泌不足为原发病理基础,属于胰岛β细胞功能遗传性缺陷的特殊类型。
MODY患病率较低,但广泛分布于欧洲、拉丁美洲、非洲及亚洲的印度和日本等人群,我国香港及台湾等地也有个别家系报道。由于疾病遗传及临床表型的高度异质性及诊断本身所存在的困难,目前还没有较完整而系统的MODY流行病学资料,各国所报道的患病率也存在较大差异,根据西欧资料统计占
糖尿病的2%~5%。
另有数据显示,被诊断为1型
糖尿病而缺乏HLA连锁的患者中约10%实际上是MODY。MODY大多病情较轻,不少患者直至中老年才获得诊断,而无症状亲属在家系调查过程中又常被忽视,因此实际的患病率可能被低估。在6种MODY亚型中,分布最广的是MODY3,几乎存在于已报道的所有人群中,约占全部MODY家系的21%~64%。其次是MODY2,为欧洲最常见类型,占8%~63%。其他亚型均较少见,如MODY5主要在日本家系中被发现,MODY 6家系目前仅报道2例。
病因
病因:随着生物学、遗传学的进展、现已证实MODY的遗传病因和单基因突变,但实变基因有遗传异质性。最早确立的MODY基因与疾病连锁关系是1991年G.L Bell等在研究RW家系中获得的。他们对该家系5代共360名家系成员进行了长达30年的回顾及追踪研究,首次发现该家族性
糖尿病与第20号染色体长臂(20q12-q13.1)上腺苷脱氨酶(adenosine deaminate,ADA)基因附近区域存在紧密连锁关系,因此将与该区域存在连锁关系的
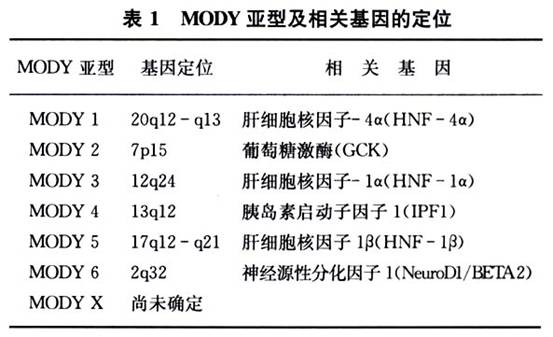
糖尿病称为MODY1。进一步研究发现,MODY1是由存在于该区域的转录因子肝细胞核因子-4α(hepatic nuclearfactor-4α,HNF-4α)基因突变引起的。随着分子生物学技术的发展和遗传统计方法的进步,以及人们对MODY高度异质性的认识,越来越多的MODY家系被研究报道及分型,目前至少已发现6种MODY亚型,除MODY1以外还包括MODY2/葡萄糖激酶(glucokinase,GCK),MODY3/肝细胞核因子1α(HNF-1α),MODY4/胰岛素启动子因子1(insulin promoter factor 1,IPF1),MODY5/肝细胞核因子1β(HNF-1β),MODY 6/神经源性分化因子1(NeuroDL/BETA2)。另外还有16%~45%的家系具有典型MODY临床表现及遗传特征,但分子遗传学机制尚未明确的,称为MODY-X(表1)。各MODY亚型除遗传背景不一样外,临床表型亦各有特点,是一组异质性的慢性高血糖综合征。
发病机制
发病机制:6种MODY亚型中,除MODY 2与葡萄糖激酶基因突变有关外,其余均为调节胰岛素基因表达的转录因子变异。葡萄糖激酶是葡萄糖酵解的第一个限速酶,在胰岛β细胞和肝细胞中催化葡萄糖转变成6-磷酸葡萄糖。胰岛β细胞中的葡萄糖激酶受血中葡萄糖浓度的调节,血糖升高可直接增强葡萄糖激酶活性,使葡萄糖代谢速度加快,进一步促进胰岛素的分泌,故胰岛β细胞中的葡萄糖激酶有“葡萄糖感受器”之称;肝脏中的葡萄糖激酶受胰岛素调节,进食后血糖升高,胰岛素分泌增多,葡萄糖激酶活性增强,促进肝细胞中糖原的合成,该酶缺陷可以导致餐后血糖升高。研究表明,β细胞葡萄糖激酶活性的轻微下降将提高葡萄糖诱导的胰岛素分泌的感受阈,这是MODY 2发生的主要机制。目前已发现130种以上与MODY 2相关的葡萄糖激酶突变,包括无义突变、错义突变、缺失突变等,这些突变通过改变酶活性及酶与葡萄糖或三磷酸腺苷的结合力,使葡萄糖激酶选择性地对血中葡萄糖浓度的“感受力”下降,胰岛素分泌的快速时相延迟或消失,分泌率下降,从而导致不适当的胰岛素分泌不足,而β细胞对于其他促胰岛素分泌物如精氨酸的刺激反应正常。此外,肝脏葡萄糖激酶的活性降低可导致6-磷酸葡萄糖的生成及肝糖原合成的速度减慢,也是引起高血糖的原因。
另外5种与转录因子变异相关的MODY亚型均以原发性胰岛素分泌缺陷而非胰岛素抵抗为病理生理基础,但具体发病机制目前仍未清楚。这些转录因子主要在肝脏、肾脏、胃肠道及胰腺细胞中表达,彼此之间形成相互调节的网络体系,共同对胚胎期胰腺发育、胰岛β细胞的增殖分化以及与葡萄糖及脂代谢相关基因的表达调控起重要作用。例如HNF-1α主要由3个功能区组成,包括氨基端的二聚化区,羧基端的反式激活区,以及中间的核酸结合区,它主要以同二聚体或与HNF-1β以异二聚体形式与所调节的相应基因片断结合;而HNF-4α是HNF-1α的上游调控因子,由于HNF-4α基因突变所导致的HNF-1α表达下降是导致葡萄糖代谢紊乱的原因之一。关于这些转录因子变异引起
糖尿病的机制,基因敲除动物模型及体外实验的结果提供了部分线索,如HNF-1α缺失的小鼠β细胞不能通过糖酵解生成还原型烟酰胺腺嘌呤二核苷酸(nicotinamide-adenine dinucleotide,NADH),进而使葡萄糖诱导的三磷酸腺苷生成减少,胰岛素分泌量降低;而功能性HNF-4α缺失的胚胎干细胞,由于受该转录因子调控的基因包括葡萄糖转运子-2、醛缩酶B、甘油醛-3-磷酸脱氢酶及丙酮酸激酶等的表达缺陷,影响了葡萄糖的转运及酵解过程,结果使胰岛素分泌受损。另外,HNF-4α还调节脂代谢途径中载脂蛋白CⅢ(apoprotein CⅢ,Apo C Ⅲ)基因的表达,研究发现某些MODY 1突变携带者其血三酰甘油和Apo C Ⅲ浓度明显低于其无突变的非
糖尿病家系成员,这可能与HNF-4α突变引起Apo C Ⅲ表达量下降,进而导致脂蛋白脂酶的活性增加和三酰甘油水平降低有关。IPF1对胚胎期胰腺的发育以及成年期胰腺内分泌特异性基因的转录调节起着重要的作用,胚胎期该蛋白的表达缺失,可导致胰腺发育不良,而其杂合子突变则通过下调相关基因的表达,从而影响了胰岛素的分泌。
目前已发现至少120种与MODY 3相关的基因突变,而由HNF-4α变异引起的MODY 1相对少见,至今全球共报道了13个家系,MODY 4~6则更少,各1~2例左右。突变包括移码突变、缺失突变、错义突变、无义突变等,一般认为,错义突变多发生在转录因子的DNA连接区及同源结构区,主要通过降低转录因子与靶基因片断的连接使下游基因表达减少;而无义突变及移码突变较少影响靶基因的连接,主要由于突变的存在,改变了反式激活区的序列,从而使该蛋白的转录功能缺失,另外突变蛋白还可通过显性负效应与野生型蛋白竞争靶基因的连接位点,进而使靶基因表达减少
临床表现
临床表现:MODY是一组以胰岛素分泌缺陷为特征的慢性高血糖综合征,其胰岛素不足程度介于1型及2型
糖尿病之间,临床表现又具有两者的某些特点,构成了
糖尿病疾病谱的中间过渡类型(表2)。
MODY的遗传异质性决定其临床表型的异质性特点。一般认为与葡萄糖激酶基因突变有关的MODY 2,因血糖升高而引起的临床表现较轻,不足一半的患者表现为显性
糖尿病。该亚型外显率较高而完全,大多数突变携带者在青春期即出现血糖水平的轻度升高,因无症状而未被发现,约50%携带突变的妇女于妊娠期通过葡萄糖耐量筛查试验发现
糖尿病,而目前通过家系调查发现的最小MODY 2患者诊断年龄为1周岁。MODY 2病情进展缓慢,许多患者可以长期保持糖耐量减低或轻度空腹高血糖。微血管并发症包括
糖尿病视网膜病变、糖尿病肾病较少见且预后良好,而与大血管并发症相关的危险因素,如高血压、
肥胖、血脂紊乱等也较少在MODY 2患者中聚集,故与之相关的心脑血管并发症也较少见。葡萄糖激酶基因突变除引起血糖增高外,还影响胚胎发育从而导致新生儿出生体重过低,这可能是胎儿期胰岛素分泌不足的结果,但这种现象在其他MODY亚型中并不多见。
由肝细胞核因子突变引起的MODY 1及MODY 3临床表现相似,外显率相对较低而不完全,高血糖发生的时间稍晚于MODY 2,60%~70%突变携带者在25岁前被诊断为
糖尿病,其余在25~60岁获得诊断,少数不外显的突变携带者终身不发生
糖尿病。这两种亚型的高血糖情况较为严重,胰岛素分泌功能以1%~4%比例逐年衰退,而胰岛素的敏感性相对正常,大多数患者体重指数较低,临床症状明显,病情随年龄加重,血糖控制情况常进行性恶化,易并发
糖尿病视网膜病变及糖尿病肾病,半数患者最终需要胰岛素治疗。少数外显率高的患者由于起病年龄小,病情较重且进展快,易被误诊为1型
糖尿病。另外由于HNF-1α还在肾脏表达,HNF-1α基因缺陷可通过改变肾远曲小管钠-葡萄糖协同转运子的表达,使肾脏重吸收葡萄糖能力下降,进而降低肾糖阈,这也是MODY 3临床表现的特点之一。
IPF1是胰腺发育及胰岛β细胞内分泌特异性基因表达的重要转录因子,目前仅发现1例因IPF1杂合子突变引起的MODY 4家系,其临床表现并不严格地符合MODY的诊断标准,如平均发病年龄较晚,约为35岁,近几年的研究发现该基因的某些位点突变似乎与晚发的普通2型
糖尿病易感性有关。HNF-1β基因突变所致的MODY 5主要在日本家系中被发现,临床上除了具有一般MODY共有的遗传特征外,大多病情较轻,可伴有肾脏先天性病变(如多囊肾)及肾功能不全,这些肾脏改变可早于高血糖的发生,部分晚期患者可能需要胰岛素治疗。与NeuroDL/BETA2突变相关的MODY 6家系仅报道2例,其中一例临床表型与MODY 3类似,而另一例则更接近于普通2型
糖尿病,即发病年龄稍晚,体型
肥胖且胰岛素分泌功能正常等。
除了上述常见的不同MODY亚型之间总体的临床表型异质性以外,同一种MODY亚型内不同家系,或同一家系的不同成员之间的临床表现也常不一致,如发病年龄的早晚及
糖尿病的严重程度等。除考虑突变类型对表型的影响外,环境因素如不同的生活方式及饮食习惯对疾病外显率的作用可能是其中的原因之一。此外,某些微效基因的突变虽然不足以导致
糖尿病的发生,但可对MODY的临床表型起修饰作用,影响了高血糖的严重程度。
治疗
治疗:MODY的治疗仍以纠正代谢紊乱、防止或延缓并发症及延长寿命为目的,因为发病年龄较小,对高血糖的控制应更为严格,控制血糖方案可根据不同MODY亚型及高血糖的严重程度决定。如MODY 2一般血糖升高较轻微,约2/3的患者可单靠控制饮食和体重,以及适当的运动,而不需要依赖药物的作用即可获得良好的血糖控制。另外1/3的患者对磺脲类降糖药有显效,除妊娠期患者外,一般无需胰岛素治疗。对于临床表现较严重的MODY亚型,如MODY 1及MODY 3则常需口服降糖药或胰岛素来控制血糖,其药物选择指征和血糖控制标准以及对并发症的监测等与普通2型糖尿病基本相似,但由于其主要病理生理机制为胰岛β细胞分泌胰岛素的不足,而胰岛素敏感性基本正常,故当口服磺脲类等促胰岛素分泌剂不能良好控制血糖水平时应尽早使用胰岛素治疗。